Entropic Driving Force Behind DNA and RNA Structures
Though the safekeeping of genetic data is maintained in the genome as a double helix, DNA must assume a transient single-stranded (ss) form during replication, transcription and a variety of repair and recombination processes. The exposed ssDNAs play a key role in these processes.
RNA are almost always single-stranded in the cell. They fold back onto themselves to assemble secondary and tertiary structures. Single-stranded regions on DNAs and RNAs can exert determinantal control over their functions.
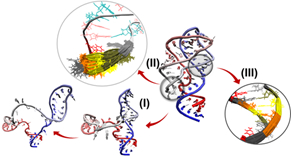
Entropic flexibilities of the nucleic acid backbone can be roughly divided into three levels according to length scales (see illustration). (I) On the coarsest level, there are “shape-shifting” fluctuations, like those involved in the folding/unfolding of an RNA or during ligand binding of a riboswitch. (II) Within the tertiary fold, there are ss tracts along the sequence, particularly in loops and junctions which are conformationally floppier than the others. (III) Finally, even within the secondary structures (e.g. helices), conformational flexibilities along the backbone remain within each nucleotide. While we may be used to seeing a single conformation in crystallographic structures, conformational fluctuations persists along the backbone. These “residual” entropic fluctuations are depicted by those motions inside the circle labeled (III).
The Mathematics of Nucleic Acid Backbone Conformations
Can the backbone of a biopolymer as complex as a nucleic acid be understood by mathematics? In a short answer: Yes!
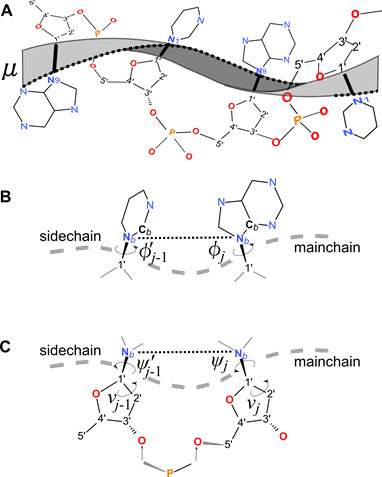
Being able to enumerate the conformational states of the sugar-phosphate chain is the key to understanding ssDNA and RNA structures. There are 30+ atoms on each nucleotide. While an analytical formulation for finding all the backbone conformations for a polymer as complicated as a nucleic acid may seem like an impossible task, there are significant redundancies on the sugar-phosphate backbone that makes the mathematics manageable. The BCV/-c solution (see figure), described in a few of our papers, consists of: (1) closure of the ribose ring, (2) the phosphate linkage closure and (3) excluded volume due to backbone atom steric interactions. The solution works for both RNA and DNA (with and without the 2’ OH).
Mapping Residual Entropic Stress Points on DNAs and RNAs
How does the folded structure of a RNA or protein-DNA interactions impart entropic strains on the backbone and how are are these distributed along the RNA or DNA?
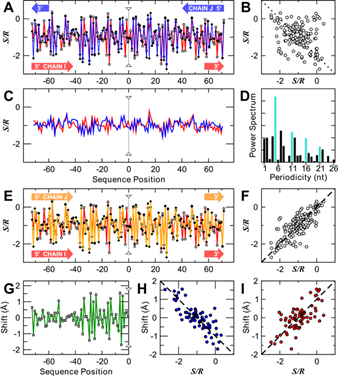
Residual entropic strains have been computed along the backbone of a number of folded RNAs and DNAs. There is a surprisingly strong correlation between level (III) entropy and structure. For example, entropic strains on nucleosomal DNAs are remarkably correlated with base pair step geometries and nucleosome positioning (right figure). Those nucleotides under highest entropy stress in the nucleosome are shown in red on the left. We found a surprisingly large free energy penalty for folding ~ 0.47 kcal/mol/nt for RNAs due to residual entropy alone, but a much milder ~ 0.09 kcal/mol/nt for DNAs. These information provide important lower bounds to the magnitudes of the other free energy determinants (e.g. stacking and pairing, and level II and III entropies) for folding and packaging.
NUCLEIC: A High-Efficiency Monte Carlo Simulation for DNAs and RNAs
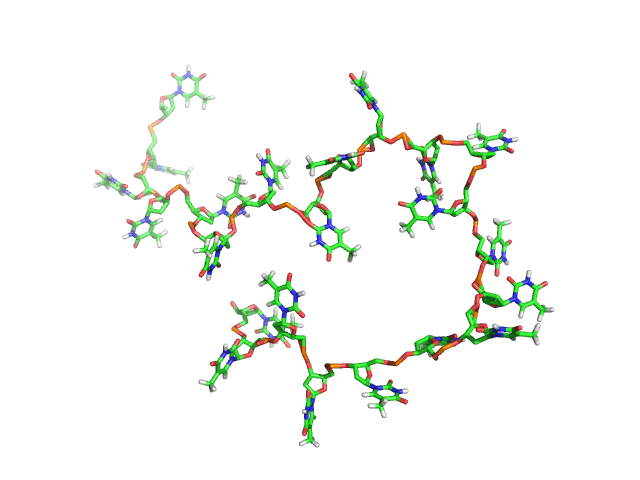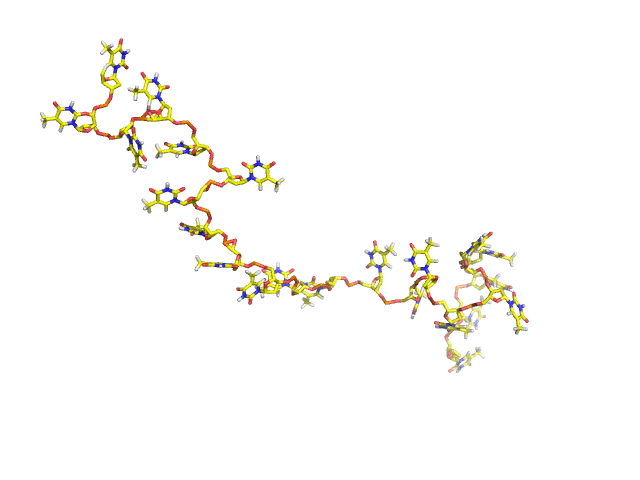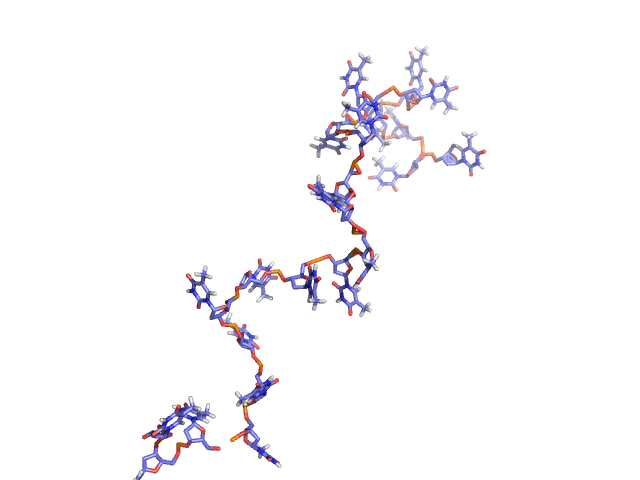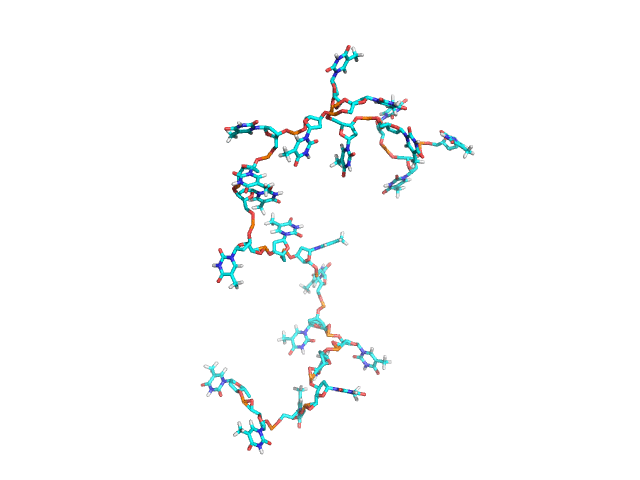
BCV/-c is the engine behind NUCLEIC, a high-efficiency Monte Carlo program for simulating DNAs and RNAs in solution. NUCLEIC is both atomistically accurate as well as predictive. NUCLEIC is strongly ergodic (MC snapshots shown on right for dT22).
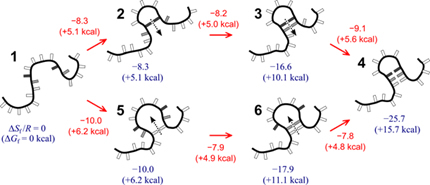
NUCLEIC not only allows us to simulate DNA and RNA structures efficiently, we can now compute the thermodynamics and kinetics of a variety of secondary structures from first principle. Left figure shows a comprehensive entropic landscape for RNA stem-loops, and values of the entropy barriers between different states during the initiation and propagation of RNA hairpins.
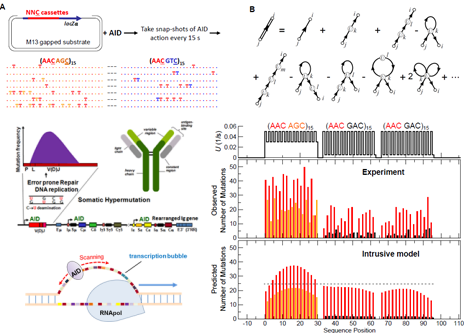
Good Mutations: Antibody Diversification Benefits from ssDNA Mutations
Antibody diversity is produced by somatic hypermutation (SHM). The enzyme AID (activation induced deoxycytidine deaminase) deposits C to U mutations randomly on the Ig gene to initiate SHM. AID acts exclusively on ssDNA during active transcription, targeting the Ig(V) region preferentially.
Stochastic modeling and path integrals can be used to compute the mutation spectrum. U mutation frequencies show a surprisingly strong dependence on the sequence of the substrate ssDNA. This has been confirmed by in vitro experiments. AID activity is a potentially powerful assay for ssDNA secondary structures.
Nucleic Acid-Ion Interactions
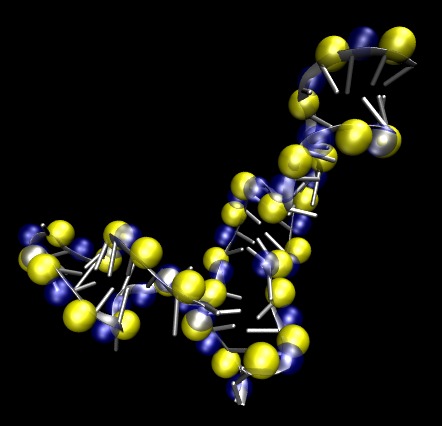
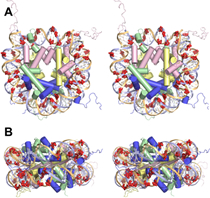
Nucleic acids (DNA and RNA) are highly charged biopolymers. Under physiological conditions, systems involving nucleic acids also contain positively-charged counterions in solution such at Mg2+ and K+. For a long time, these counterions were thought to act merely as the enforcers of charge neutrality. It is now clear that counterions serve a much larger function. Counterions can stabilize the folding of RNA, can mediate the effective attractive interactions of DNAs during packaging and can modulate peptide-DNA and peptide-RNA interactions. The picture to the left shows how Mg2+ ions (yellow) are typically correlated with the backbone of a RNA under physiological salt conditions (~ 1 mM Mg2+).

New Algorithms for Large-Scale Computer Simulations of Complex Systems
We continue to engage in the fundamental development of simulation algorithms that would enable chemists and molecular biologists to carry out calculations for large-scale and complex classical and quantum systems. Some of these new algorithms are already deployed in the studies of nanoscale superfluidity and RNA folding. Other projects currently being developed include real-time path integral simulations for condensed-phase quantum dynamics, as well as simulations using the stochastic potential switching (SPS) algorithm to study biological systems and complex fluids. The movie to the right shows an example revealing the intricate particle correlations in 1/4 x 1/4 portion (about 65,000 particles in each frame) of a 1-million particle simulation, studying the melting of a 2-dimensional fluid. This study with more than 4 million particles, carried out back in 2006, still holds the world's record on the largest 2-d melting simulation ever!